
by Emily Hasser | Dec 22, 2020 | Disease Genetics, Evolution
Main Takeaways:
- Genetic mutations that lead to uncontrolled cell proliferation can cause cancerous tumors. Random genetic mutations occur at every cell division. As more cell divisions accumulate, there are increased chances for a cancer-causing mutation. While one would expect that larger organisms would be at an increased risk for cancer because they accumulate greater amounts of cell divisions, this is not the case for elephants and whales.
- Several hypotheses may explain this paradox: shorter telomeres, negative selection against hypertumors, and variation in the TP53 gene. Read below for more detail.
In the United States, the National Cancer Institute estimates that about 1.8 million new cancer diagnoses and approximately 600,000 cancer-related deaths will occur in 2020 [1]. Furthermore, about 39.5% of people can expect to receive a cancer diagnosis at some point in their life [1]. Although cancer has many possible causes, downstream genetic mutations ultimately drive the development of cancer [2].
In multicellular organisms, genetic mutations don’t just affect a single, isolated cell, but can be passed on to many cells in the organism through cell division as the organism grows. Random genetic mutations occur at every cell division, and some of these can lead to uncontrolled cell proliferation resulting in the growth of tumors that can become cancerous. As an organism grows larger and lives longer, more cell divisions will occur in its body over its lifetime. One might expect that larger organisms would accumulate more mutations due to increased numbers of cell divisions and that there should be more chances for tumor growth. However, mortality due to cancer in large animals with long lifespans is not higher than in humans: this discrepancy is known as Peto’s Paradox [3]. If organisms the size of whales had up to 1,000 times the cancer risk than humans, as we might expect given the increased number of cell divisions and random mutations, it would be very unlikely for them to reproduce before succumbing to cancer.
Several hypotheses have been proposed to account for Peto’s Paradox, using data collected from large animals such as whales and elephants [3,4]. Studying cancer in wild mammals presents unique challenges due the lack of accessibility to populations and inability to control factors such as environmental exposures. However, whales, porpoises, and dolphins are all cetaceans, which is a group of wild animals that has been extensively studied [4]. Except for a few cases linked to environmental pollution, cancer in whales rarely occurs [3,4]. This makes them a great model organism for studying Peto’s Paradox. Since these organisms do not appear to have a high risk of cancer, it is suggested that their cancer prevention mechanisms are likely more effective than those of smaller organisms. Other researchers maintain that the rate of cancer development is the same, but cancer may not be as lethal in larger organisms. Here, I will describe hypotheses explaining Peto’s Paradox involving telomeres, hypertumors, and the tumor suppressor gene p53, in the order of least to most investigated.
Telomeres
Telomeres may play a critical role in cancer suppression in large organisms. Similar to how shoelaces have plastic aglets to prevent fraying, telomeres are repetitive DNA sequences located on both ends of each chromosome to prevent DNA damage [5]. Telomeres protect the important stretches of the genome located in the middle of the chromosomes and have recently become a hot topic in research due to possible implications in aging and cancer [5].

Telomeres (pictured in purple) gradually shorten each time a cell divides.
Telomeres deplete with every round of DNA replication. When the telomeres become too short, the cell becomes senescent, entering a dormant state in which it doesn’t divide. Researchers hypothesize that shortened telomeres in large organisms with long lifespans could explain their reduced cancer incidence [3]. Reducing the number of times an individual cell divides would reduce the opportunities for a mutation in an oncogene or tumor suppressor gene to occur. With fewer previous divisions for mutations to accumulate, the cell has a decreased risk of developing cancer. The exploration of telomere modifications as an explanation for Peto’s Paradox has just begun, and further research is needed to investigate the possibility.
Hypertumors
Another new hypothesis proposed to explain Peto’s Paradox is promising, but has only been illustrated in silico, or through a computer simulation. Researchers suggest that in larger animals, malignant tumors have a fitness disadvantage compared to benign tumors [4]. In a population of cancer cells with various phenotypes, natural selection may favor aggressive “hypertumors” that piggyback off the vascular growth of parent tumors. Acting as parasites, these hypertumors deplete the parent tumor’s resources and eventually destroy it. Unlike in small organisms, tumors need to reach a substantial size to have consequences in large organisms. Therefore, hypertumors have plenty of time to develop and damage the original tumor before the original tumor grows to a lethal size. As a result, cancer may still be more common in large organisms, just less lethal [4]. Additional research investigating tumor growth in living whales is needed before any concrete conclusions can be drawn.
TP53
Variation in TP53 has been identified as another possible explanation for Peto’s Paradox [3,6]. As a tumor suppressor gene, TP53 helps control cell growth, and mutations in the TP53 gene have been found in up to 50% of human cancers. The p53 protein expressed by this gene has primary roles in cell cycle arrest, DNA repair, and apoptosis. Mutations in the TP53 can lead to reduced expression of p53 and the uncontrolled cell growth that is a hallmark of cancer [7].
Imagine the cell cycle as the process of loading laundry into your washing machine. You’ve put the clothes and detergent in and started the wash cycle when soapy water suddenly starts seeping out of the crevices. Your instinct in this situation is probably to stop and turn off the washing machine to investigate the problem. Likewise, cell cycle arrest occurs when the cell notices that something is wrong, and all duplication processes stop to identify the problem. If you notice a fraying, broken wire poking out from behind the wall, you will probably choose to call an electrician instead of risking a do-it-yourself fix. In the cell cycle, p53 recognizes DNA damage and activates DNA damage response pathways to initiate DNA repair. Sometimes, your machine might be too broken to fix, and you must resort to removing it from your house and heading out to buy a new one. In a cell, the analogous process is apoptosis, where a severely damaged cell is marked for destruction and effectively killed before it can do further damage or proliferate.
To investigate the relationship of p53 and Peto’s Paradox, genome-wide studies were conducted, synthesizing data from 61 animals with a range of sizes [6]. These animals included large species such as the Asian elephant and woolly mammoth. Researchers found that as species evolved to be bigger, they acquired more copies of TP53, the gene that encodes the p53 protein. While humans only have one copy of the TP53 gene, the elephant genome contains 20 copies of TP53, resulting in greater production of p53 protein. This increase may be responsible for protecting these large organisms from developing cancer [6,8]. The role of p53 has evolutionary implications because when p53 evolved as a way to regulate the cell cycle, it fortified the system in place to ensure DNA was replicated correctly and cells with mistakes were killed before the issue could spread.
Although many explanations have been proposed to account for Peto’s Paradox, more research is necessary to elucidate the precise molecular basis of the paradox. Mouse models currently dominate studies in cancer research, but they are small organisms with short life spans. Although these characteristics are useful when researchers want to conduct a study over an organism’s lifetime, they also mean that the mouse model is not the ideal model for studying cancer suppression. Expanding the study of cancer to a more diverse variety of organisms would allow for a more complete understanding of the underlying mechanisms.
Understanding Peto’s Paradox is not only important for conservation scientists and wildlife zoologists; it also has implications in human medicine. Peto’s Paradox provides an interesting opportunity for potential research that may provide insight into cancer treatment. Determining the mechanism behind cancer resistance in large animals could reveal powerful techniques to develop novel treatments for human cancers by targeting telomere length, p53 expression, or hypertumors.
References
[1] Cancer Statistics. National Cancer Institute. https://www.cancer.gov/about-cancer/understanding/statistics. Accessed December 20, 2020.
[2] Griffiths AJF, Miller JH, Suzuki DT, et al. An Introduction to Genetic Analysis. 7th edition. New York: W. H. Freeman; 2000. Mutation and cancer. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21809/
[3] Caulin AF, Maley CC. Peto’s Paradox: evolution’s prescription for cancer prevention. Trends in Ecology & Evolution. 2011;26(4):175-182. doi:10.1016/j.tree.2011.01.002
[4] Nagy JD, Victor EM, Cropper JH. Why don’t all whales have cancer? A novel hypothesis resolving Peto’s paradox. Integrative and Comparative Biology. 2007;47(2):317-328. doi:10.1093/icb/icm062
[5] Are Telomeres the Key to Aging and Cancer. Learn.Genetics Genetic Science Learning Center. https://learn.genetics.utah.edu/content/basics/telomeres/. Accessed November 15, 2020.
[6] Sulak M, Fong L, Mika K, et al. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. eLife. 2016;5. doi:10.7554/elife.11994
[7] Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. doi:10.1101/cshperspect.a001008
[8] Abegglen LM, Caulin AF, Chan A, et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA. 2015;314(17):1850-1860. doi:10.1001/jama.2015.13134

by Diane Xue | Jun 1, 2020 | Disease Genetics, Genetic Misconceptions
Main Takeaways:
- Late-onset Alzheimer’s Disease (LOAD) is influenced by many genetic and environmental risk factors. There is no gene that guarantees onset or immunity from (LOAD).
- At-home genetic testing companies that offer an “Alzheimer’s test” are only testing one variant from one gene (APOE). The presence or absence of this APOEε4 variant is not a diagnosis.
- Thus far, over two dozen risk loci for LOAD have been discovered, yet a genetic test is not sufficient to detect one’s true risk of developing disease.
When I was seven years old, my parents took me back to their hometown of Shanghai to visit my grandparents. It was a long trip we had taken before, and while the 18-hour journey was tiring, I was looking forward to seeing family, eating street food, and finding cats in the park. This trip was different, however. My mom explained to me that my grandpa may not remember us. He has Alzheimer’s, she said. Although I didn’t know what that meant, it made sense to me that my grandpa may not remember me. After all, I had changed a lot between three and seven. When we finally arrived at my grandparents’ home, I realized that it wasn’t that my grandpa was forgetful; there was a blankness. He couldn’t leave his bed.
My grandpa was suffering from a neurodegenerative disease – meaning it affects the brain and becomes worse over time. Alzheimer’s disease is the most common cause of dementia, affecting an estimated 5.8 million people in the United States and 1 out of every 10 people over age 65 [1]. The disease is devastating for those affected, their caregivers, and their loved ones. When 23andMe and other at-home (direct-to-consumer) genetic testing companies began testing for Alzheimer’s disease risk, I understood the impulse to spit in a tube and find out one’s genetic risk for the disease [2]. That impulse is driven by the feeling of fear that immediately follows annoyance when my mom turns the car back around two minutes after leaving the house because she can’t remember if she left the stove on. Of course, I want to know my risk for disease and everything I can do to prevent it. The fact is, that’s just not possible yet.
That impulse to order the genetic test is also based on the misconception that all of the genetic causes of Alzheimer’s disease are known. Last year Cosmopolitan published an op-ed titled “I tested positive for the Alzheimer’s Gene at 26 years old,” where the author shared her emotional experiences after receiving her genetic testing results and the lifestyle changes she made to help prevent the disease [3]. While I don’t believe genetic testing companies are trying to be scientifically deceiving, the op-ed highlights the consequences and unwarranted anxiety that can occur when the science and genetics of Alzheimer’s disease are not clearly presented. In this post, I will share the facts about Alzheimer’s Disease genetics. (For the rest of this post, Alzheimer’s Disease will be referred to as AD).
There are two classifications of AD: early-onset AD and late-onset AD. Early-onset AD is rare. It accounts for about 1% of all AD cases [4]. Unlike most AD cases, the genetic causes of early-onset AD are known; it is caused by mutations in the genes APP, PSEN1, or PSEN2. Early-onset AD is transmitted in a dominant Mendelian pattern. For every gene, you receive one copy (allele) from your father and one from your mother. A dominant Mendelian pattern means that if you receive just one copy with a mutation, you will develop the disease. Once again, I want to reiterate that this type of AD is very rare. If you have a family history of early-onset AD and are not in contact with a medical provider or research group, you can find more information and resources here [5].
The more common version of AD, the type that my grandpa had and the version most direct-to-consumer genetic testing companies offer a test for, is late-onset AD. Unlike the early-onset version, late-onset AD is a complex, genetically heterogeneous trait, meaning it is influenced by both environmental and genetic factors. Furthermore, different genetic variants cause the disease in different people. In other words, there is no “Alzheimer’s gene.” There are many variants in many genes that add to one’s genetic risk, and many risk variants have not been discovered yet.
So why the hype? What is the scientific basis for the direct-to-consumer late-onset AD tests that have resulted in online forums, Facebook support groups, and op-eds in popular magazines?
The direct-to-consumer tests calculate genetic risk based on the variation in a single gene: APOE. Everyone has the APOE gene on chromosome 19; what matters for AD risk is which variants of the gene you have [6]. There are three variants (six possible combinations): ε2, ε3, and ε4. APOE ε2 has been found to have protective effects, while APOE ε4 is the variant associated with an increased risk of late-onset AD. Studies have found that on average, having one copy of ε4 increases the risk for late-onset AD by 3-fold, and having two APOE ε4 alleles increases risk by 12-fold [4].
I am not trying to dismiss concerns over this increased risk; three times higher and twelve times higher risk are significant. APOE ε4 is the strongest genetic risk factor for late-onset AD that we know of. However, it is important to keep the following in mind:
(1) Inheriting two copies of APOE ε4 is rare, affecting only 2-3% of the population (see image below) [4].
(2) Late-onset AD is influenced by genetic risk factors AND environmental factors, with one study showing that 47% of the variance of AD is due to non-genetic factors [7].
(3) Having two copies of APOE ε4 is not an Alzheimer’s disease diagnosis, nor is the absence of APOE ε4 a guarantee that you will not get AD. Environmental factors aside, there are many other genetic variants that confer risk for late-onset AD (see image below).
(4) Finally, the overwhelming majority (over 90%) of research on the genetic risks of Alzheimer’s have been conducted in samples of people of non-Hispanic European descent [8]. The few studies of APOE risk on AD in diverse populations have shown that the risk of AD caused by the APOE ε4 allele varies depending on ancestry. In 2019, a study of late-onset AD in Caribbean Hispanics found that individuals with African-derived ε4 allele at the APOE gene had 39% lower odds of having AD than those with a European-ancestry-derived copy of ε4 [9].
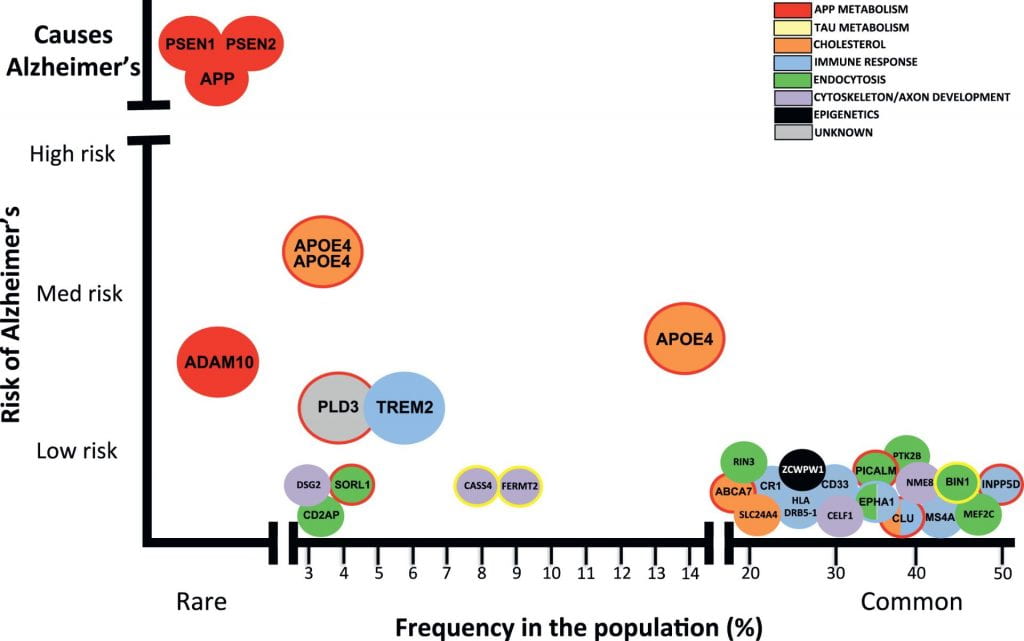
Image from Celeste M. Karch, and Alison M. Goate, “Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis.” Alzheimer’s risk-variants are plotted with frequency in the population on the X-axis and the risk conferred by each variant on the Y-axis. Mutations in PSEN1, PSEN2, and APP cause early-onset Alzheimer’s disease. Dozens of genetic mutations have been confirmed to increase risk of late-onset Alzheimer’s disease, with APOE ε4 being the strongest known disease-variant.
The bottom line is that these direct-to-consumer genetic tests of late-onset AD can tell you one piece about your risk for disease, but it can’t factor in how APOE interacts with other genes or with your environment; the test is based primarily on research of those with European ancestry; and it cannot tell you whether or not you will have Alzheimer’s disease. I look forward to the day when I can edit this post to say that an accurate assessment for late-onset AD genetic risk exists, but we’re not quite there yet. (note: if you are thinking about ordering an direct-to-consumer genetic test for AD, the Alzheimer’s Association strongly recommends you consult with a genetic counselor before the test and after receiving your results).
That trip in 2004 was the last time I saw my grandpa. At the time, I didn’t know what was happening, but I could feel the hopelessness in the air. It hits kids hard when the adults in the room are saying they don’t know anything more, that nothing can be done. I understand that it can seem discouraging that there is so much we still don’t know, that there is still no cure. But I am not hopeless anymore. In 2004, studies of the human genome were in its infancy, with The Human Genome Project having been completed only months before. At the time, there had been no genome-wide studies of Alzheimer’s disease. In the past 15 years alone, 40 genomic regions that are associated with late-onset AD have been found [10]. Every year, we learn more about new risk variants and biological pathways that can be targeted for pharmaceutical therapies. Significant efforts are being put toward researching the genetics of Alzheimer’s, worldwide, with the hope that one day we will understand the entire genetic architecture of the disease. I am optimistic for what the next 15 years will bring.
References:
[1] 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Association. Retrieved from https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf
[2] Late-onset Alzheimer’s Disease. 23andMe. Retrieved from https://www.23andme.com/topics/health-predispositions/late-onset-alzheimers/
[3] Brown, S. “I Tested Positive for the Alzheimer’s Gene at 26 years old.” Cosmopolitan Sep. 19, 2019. Retrieved from https://www.cosmopolitan.com/health-fitness/a29107622/alzheimers-gene/
[4] Karch, Celeste M. and Alison M. Goate. 2015. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Vol. 77. doi:https://doi.org/10.1016/j.biopsych.2014.05.006. http://www.sciencedirect.com/science/article/pii/S0006322314003394.
[5] Younger/Early-Onset Alzheimer’s. Alzheimer’s Association. Retrieved from https://www.alz.org/alzheimers-dementia/what-is-alzheimers/younger-early-onset
[6] Roses, Allen D. and Ann M. Saunders. 1994. APOE is a Major Susceptibility Gene for Alzheimer’s Disease. Vol. 5. doi:https://doi.org/10.1016/0958-1669(94)90091-4. http://www.sciencedirect.com/science/article/pii/0958166994900914.
[7] Ridge, PG, Hoyt, KB, Boehme, K et al. 2016. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. Vol. 41 doi.org/10.1016/j.neurobiolaging.2016.02.024.
[8] Popejoy, AB, and Fullerton, SM. 2016. Genomics is failing on diversity. Nature, 538(7624), 161–164. https://doi.org/10.1038/538161a
[9] Blue EE, Horimoto ARVR, Mukherjee S, Wijsman EM, Thornton TA. 2019. Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimer’s & Dementia: the Journal of the Alzheimer’s Association. Vol. 12. doi.org/10.1016/j.jalz.2019.07.016
[10] Andrews, Shea J., Brian Fulton-Howard, and Alison Goate. 2020. Interpretation of Risk Loci from Genome-Wide Association Studies of Alzheimer’s Disease. Vol. 19. doi:https://doi.org/10.1016/S1474-4422(19)30435-1. http://www.sciencedirect.com/science/article/pii/S1474442219304351.

Recent Comments